Um phylogenetische Bäume rekonstruieren zu können, sind wir auf Datenmaterial angewiesen, das die Entwicklungsgeschichte der Organismen widerspiegelt, d.h. in diesen Daten müssen die Spuren des Entwicklungsprozesses erkennbar sein. Früher wurden zur Rekonstruktion von Stammbäumen in erster Linie morphologische Daten herangezogen.
Eine andere Art von Daten, die vor allem in den letzten 15 Jahren vermehrt für Stammbaumanalysen herangezogen wird, sind molekulargenetische Sequenzdaten von Makromolekülen, wie DNA und Proteinen. (Futuyma, 1986)
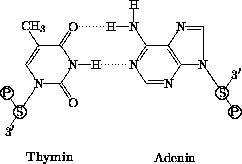
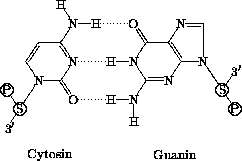
Die genetische Information liegt in der Zelle in Form langer DNA-Ketten (Desoxyribonucleinsäure-Ketten) vor. Diese Ketten setzen sich aus Nucleotiden zusammen, die mit unterschiedlichen organischen Basen beladen und über eine Phosphodiesterbrücke zwischen der 3'-OH-Gruppe und der 5'-OH-Gruppe ihrer Desoxyribose miteinander verbunden sind. Die gebundenen Nucleotidbasen gliedern sich in zwei Typen: die Purine Adenin und Guanin sowie die Pyrimidine Cytosin und Thymin.
Die Nucleinsäuren haben die Eigenschaft, miteinander
Basenpaarungen eingehen zu können, indem sie untereinander
Wasserstoffbrückenbindungen ausbilden. Hierbei können sich Adenin
mit Thymin und Guanin mit Cytosin paaren
(Abb. 1.2 und 1.3).
Seit der Erfindung von Sequenziermethoden, wie der Sequenzierung nach Sanger et al. (1977), ist es möglich, die Reihenfolgen der Basen auf molekularer Ebene zu entschlüsseln (zu sequenzieren). Diese Information liegt auf der DNA gerichtet vor. Daher kann der Inhalt, der durch die oben genannten Sequenziermethoden entschlüsselt wurde, als Zeichenketten der Buchstaben A, C, G und T (Adenin, Cytosin, Guanin und Thymin) vom 5'- zum 3'-Ende der DNA-Sequenz betrachtet werden. Informatisch gesehen, können Gensequenzen als Worte über einem Alphabet, bestehend aus den vier Symbolen A, C, G und T, dargestellt werden. (Klaeren, 1991; Li and Graur, 1991)
Seit der Erfindung der oben genannten Sequenziermethoden und deren Automatisierung durch Sequenziermaschinen, wie ALF von Pharmacia (Ansorge et al., 1992, 1993) und ABI 373A (de Bellis et al., 1994), wird eine immer größere Anzahl von Sequenzdaten immer schneller verfügbar. Dieses Anwachsen wird durch die große Anzahl an Genom-Projekten, wie z.B. Drosophila Genome Project, Yeast Genome Projekt und Human Genome Project, noch zusätzlich forciert (Ajioka et al., 1991; Goffeau and Vassarotti, 1991; Watson, 1990). Da diese Daten im allgemeinen in großen Gendatenbanken, z.B. der EMBL/EBI-Datenbank (Emmert et al., 1994; Rice et al., 1993) und GenBank (Benson et al., 1994) frei verfügbar sind, deren Inhalte exponentiell wachsen, erhalten wir die Möglichkeit, die Evolution auf molekularbiologischer Ebene zu betrachten.
Der Vorteil molekulargenetischer Sequenzdaten gegenüber morphologischen Merkmalen liegt darin, daß jede Base in einer Gensequenz oder jede Aminosäure in einem Protein als einzeln untersuchbares Merkmal betrachtet werden kann. Somit können die vorliegenden Sequenzdaten im Vergleich zu den morphologischen Daten einen größeren Merkmalsumfang und so eine feinere Auflösung der Untersuchungsergebnisse liefern. (Futuyma, 1986)