Es ist natürlich nicht klar, daß an einem Verzweigungspunkt immer genau zwei Arten entstanden sind. Diese Ungenauigkeit wird dadurch umgangen, daß man Kanten mit einer Länge null zuläßt. Dies bedeutet, daß die beiden adjazenten Knoten der ,,Nullkante`` denselben Vorfahren repräsentieren. Beide Knoten haben dann dieselben Merkmale, da die Evolution keine Zeit hatte, eines der Merkmale zu verändern. (Felsenstein, 1981)
![\begin{figure}
\centering
\hfill
\subfigure[]{
\beginpicture \scriptsize
\setco...
...5 48
\setquadratic
\plot 35 10 42.5 6 50 10 /
\endpicture}
\hfill~\end{figure}](img13.gif) |
Bei der Rekonstruktion phylogenetischer Stammbäume werden im Idealfall alle möglichen Bäume anhand eines vorgegebenen Kriteriums (z.B. Maximum Likelihood) untersucht, um sicher zu gehen, daß man auch den nach diesem Kriterium besten Baum findet. (Felsenstein, 1981, 1982; Swofford and Olsen, 1990)
Hierzu muß man allerdings wissen, daß für n Spezies
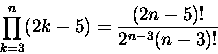
Aufgrund dieses immensen Wachstums der Anzahl der Bäume,
ist es im allgemeinen nicht möglich, die Gesamtheit aller
möglichen Bäume zu betrachten, um den optimalen Baum zu finden.
Aus diesem Grund werden heuristische Verfahren verwendet, um die
Anzahl der zu betrachtenden Bäume einzuschränken.
Zwei häufig verwendete Verfahren sind das
Clustering-Verfahren![[*]](foot_motif.gif) und das schrittweise Einfügen (Swofford and Olsen, 1990).
und das schrittweise Einfügen (Swofford and Olsen, 1990).
Beim Clustering werden die Sequenzen oder Sequenzgruppen gesucht, die sich nach einem bestimmten Maß, meist der evolutionären Distanz zwischen den Sequenzen, am ähnlichsten sind. Diese bilden zusammen eine neue Gruppe und gehen als solche in das weitere Clustering ein. (Swofford and Olsen, 1990)
Beim schrittweisen Einfügen wird, angefangen mit einer kleinen Teilmenge der zu untersuchenden Taxa, der optimale Baum für diese Taxa bestimmt. Anschließend werden Schritt für Schritt weitere Taxa eingefügt und der, basierend auf dem vorher gefundenen Baum, nächste optimale Baum gesucht. (Felsenstein, 1981; Swofford and Olsen, 1990)
Bei beiden oben genannten Verfahren handelt es sich um
Greedy-Verfahren,
die sich Schritt für Schritt von einer lokal besten Entscheidung
zur nächsten bewegen.
Diese Verfahren sich vergleichbar mit den in der
Informatik verwendeten Verfahren von Kruskal bzw. Prim
zum Berechnen Minimaler Spannbäume (MSB) in Graphen
(, )[persönliche Kommunikation]vingron-mitteilung.
Clustering entspricht hierbei der Methode von Kruskal, bei der die kürzeste verbindende Kante zwischen zwei Zusammenhangskomponenten im Graphen gesucht wird, die dann durch die gemeinsame Zusammenhangskomponente, hier Organismen-Gruppen, ersetzt werden. Dieses wird solange fortgeführt, bis alle Knoten des Graphen zu einer Zusammenhangskomponente verbunden sind (Cormen et al., 1990; Ottmann and Widmayer, 1993).
Prim's Algorithmus entspricht dem schrittweisen Einfügen. Hierbei wird die kürzeste Kante gesucht, die einen neuen Knoten mit einer bestehenden Zusammenhangskomponente verbindet. Dabei wird nur eine Zusammenhangskomponente, hier Bäume der bisher betrachteten Organismen, schrittweise vergrößert, bis alle Knoten enthalten sind (Cormen et al., 1990; Ottmann and Widmayer, 1993).
![\begin{figure}
\begin{displaymath}
\beginpicture \scriptsize
\setcoordinatesyste...
...*{2.1}} [Bl] at 35 10
\put {R} at 35 7
\endpicture\end{displaymath} \end{figure}](img11.gif)