_________ ___ ____ _____
/ ___/ __ \/ _ \/ __ \/ ___/
/ /__/ /_/ / // / /_/ / /__
\___/\____/____/\____/\___/
CODOC is a compressed data format and API for coverage data stemming from sequencing experiments
About
CODOC is an open file format and API for the lossless and
lossy compression of depth-of-coverage (DOC) signals stemming from
high-throughput sequencing (HTS) experiments. DOC data is a
one-dimensional signal representing the number of reads covering
each reference nucleotide in a HTS data set. DOC is highly-valuable
information for interpreting (e.g., RNA-seq), filtering (e.g., SNP
analysis) or detecting novel features (e.g., structural variants) in
sequencing data sets.
If you make use of CODOC, please cite:
Niko Popitsch,
CODOC: efficient access, analysis and compression of depth of coverage signals.,
Bioinformatics, 2014,
doi:10.1093/bioinformatics/btu362
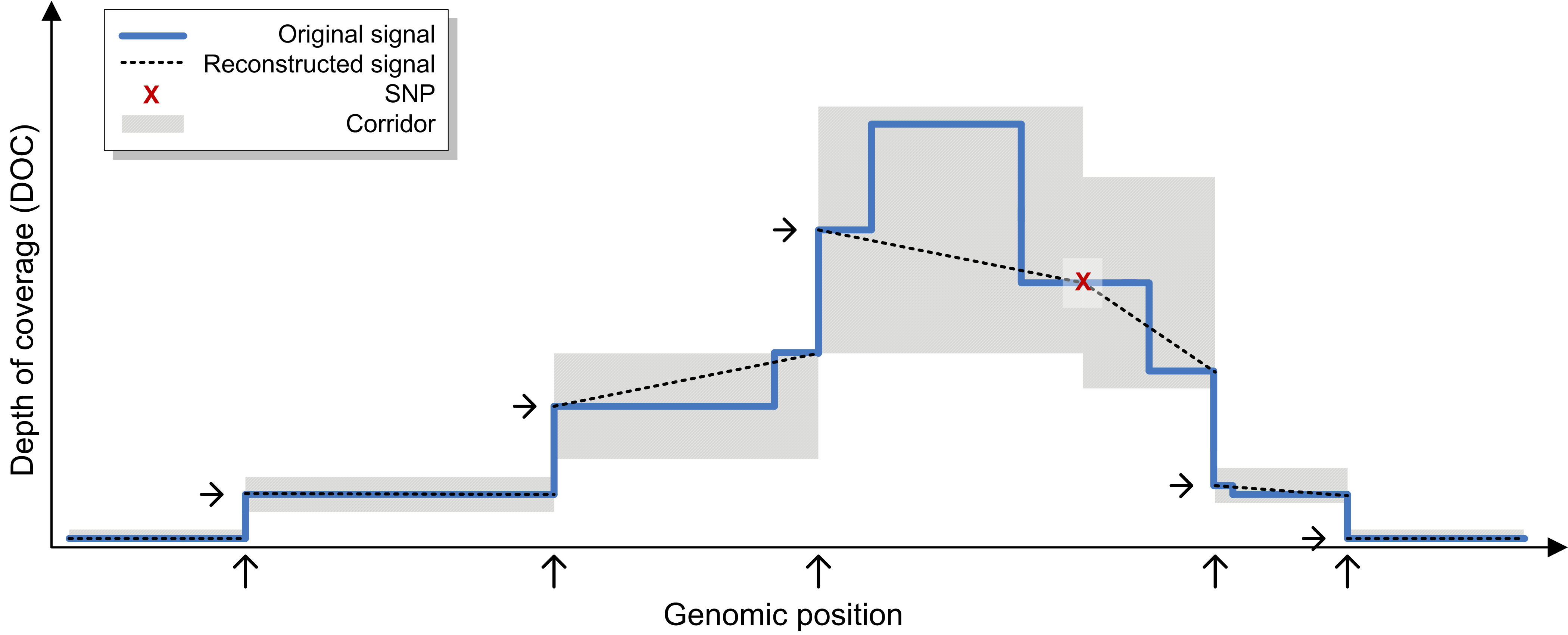 CODOC exploits
several characteristics of DOC data stemming from HTS data and uses
a non-uniform quantization model that preserves DOC signals in
low-coverage regions nearly perfectly while allowing more divergence
in highly covered regions.
CODOC exploits
several characteristics of DOC data stemming from HTS data and uses
a non-uniform quantization model that preserves DOC signals in
low-coverage regions nearly perfectly while allowing more divergence
in highly covered regions.
CODOC reduces required file sizes for DOC data up to 3500X when compared to raw representations and about 4-32X when compared to the other methods. The CODOC API supports efficient sequential and random access to compressed data sets.
Some usage scenarios for CODOC:- extract strand-specific coverage signal (export as WIG file)
- query coverage values at given genomic positions
- extract genomic regions with a given minimum/maximum coverage
- filter single-nucleotide variations in cancer/normal pairs
- pairwise combinations of coverage files (e.g., subtract one signal from another, calculate minimum signal, etc.)
- cross-correlation of coverage signals
- compression of DOC data (e.g, for archiving or transmission). Note that successive compression/decompression of DOC data with CODOC can be used for intentional data quantization.
Getting CODOC
CODOC source code and releases can be downloaded from GitHub. CODOC uses maven as build tool. For development we recommend the Eclipse IDE for Java developers and the m2e Maven Integration for Eclipse.
The CODOC jars can be built with
bin/build-java.sh <VERSION>
(version is, e.g., 0.0.1)
Usage Examples
Call the CODOC API w/o parameters to get short usage information (see below) for the individual commands:
$ java -jar bin/codoc-0.0.1.jar
$ java -jar bin/codoc-0.0.1.jar compress
$ java -jar bin/codoc-0.0.1.jar decompress
$ java -jar bin/codoc-0.0.1.jar tools
The following command compresses coverage data extracted from a SAM/BAM
file using a regular grid of height 5 for quantization. Only reads
that have the SAM flag for PCR or optical duplicates (1024) unset and
reads with a mapping quality > 20 are included.
$ java -jar bin/codoc-0.0.1.jar compress -bam BAM -qmethod GRID -qparam 5 -filter "FLAGS^^1024" -filter "MAPQ>20" -o BAM.comp
The following command compresses coverage data extracted from all minus-strand reads in
a BAM file losslessly. Only reads with a mapping quality > 20 are included.
$ java -jar bin/codoc-0.0.1.jar compress -bam BAM -qparam 0 -filter "STRAND=-" -filter "MAPQ>20" -o BAM.comp
The following command prints the header of a compressed file
showing the metadata as well as the configuration used for
compressing the file.
$ java -jar bin/codoc-0.0.1.jar decompress head -covFile BAM.comp
The following command starts an interactive query session for random accessing the
compressed file.
$ java -jar bin/codoc-0.0.1.jar decompress query -covFile BAM.comp
The following command extracts all regions from the compressed file
that have a coverage between MIN and MAX and stores them to a BED file. The given name/description are used
for the BED track.
$ java -jar bin/codoc-0.0.1.jar decompress tobed -covFile BAM.comp -outFile BED -min MIN -max MAX -name NAME -description DESCRIPTION
The following command queries a file and scales the results by a linear factor.
$ java -jar codoc-0.0.1.jar decompress query -covFile BAM.comp -scale 2.0
$ java -jar codoc-0.0.1.jar decompress towig -covFile BAM.comp -o BAM.comp.wig
$ wigToBigWig BAM.comp.wig chrSizes BAM.comp.bw
java -Xmx4g ...
Usage
Usage of the compressor:
Usage: java -jar x.jar compress [-bedFile <arg>] [-best] [-blockSize <arg>] [-compressionAlgorithm <arg>] -cov <arg>
[-createStats] [-dumpRawCoverage] [-filter <arg>] [-keepWorkDir] [-manualGolombK <arg>] -o <arg> [-qmethod <arg>]
[-qparam <arg>] [-scaleCoverage <arg>] [-statsFile <arg>] [-t <arg>] [-v] [-vcf <arg>]
Mandatory Params
-cov,--covFile <arg> Input coverage file. Supported file types are SAM, BAM, WIG and bedtools coverage files.
-o,--out <arg> Output file
Params
-bedFile <arg> Input BED file containing regions of interest (optional). The file will be interpreted 0-based!
-best Used best (bzip2) compression method. Compression/decompression times might be slighly slower with this method.
-blockSize <arg> Number of codewords per data block (default: 100000)
-compressionAlgorithm <arg> Used envelope compression algorithm [NONE, GZIP, BZIP2]. Default method is GZIP
-cov,--covFile <arg> Input coverage file. Supported file types are SAM, BAM, WIG and bedtools coverage files.
-createStats Create a statistics file (default: false)
-dumpRawCoverage Create raw coverage file (<outfile>.rawcoverage). No heading or trailing 0-coverage zones will be included. Output will have 1-based coordinates.
-filter <arg> Filter used reads by SAM attribute (applicable only in combination with SAM/BAM files). Multiple filters can be provided that will be combined by a logical AND. Note that filters have no effect on reads that have no value for the according attribute. Examples: 'X0>9', 'X1=ABC', 'STRAND=+''FLAGS^^512', 'none',... [default: -filter FLAGS^^1024 -filter FLAGS^^512].See below for more help on filters.
-keepWorkDir Do not delete the work directory (e.g., for debugging purposes)
-manualGolombK <arg> Optional parameter for manual determination of the Golomb-encoding k parameter (if unset, it will be estimated from the data). (default: none)
-o,--out <arg> Output file
-qmethod <arg> Quantization method (PERC, GRID, LOG2); default: PERC
-qparam <arg> Quantization parameter (default: 0.2)
-scaleCoverage <arg> Optional parameter for scaling the coverage values. Applicable only for coverage file input (not for BAM files). (default: none)
-statsFile <arg> Statistics file name (default: <outfile>.stats)
-t,--temp <arg> Temporary working directory (default is current dir)
-v,--verbose be verbose
-vcf,--vcfFile <arg> Input VCF file (optional)
-------------------------------- FILTERS -----------------------------------
Filters can be used to restrict the coverage extraction from am SAM/BAM file
to reads that match the given filter criteria. Multiple filters are combined
by a logical AND. Filters are of the form <FIELD><OPERATOR><VALUE>.
Possible fields:
MAPQ the mapping quality
FLAGS the read flags
STRAND the read strand (+/-)
FOPSTRAND the first-of-pair read strand (+/-)
Other names will be mapped directly to the optional field name in the SAM file.
Use e.g., NM for the 'number of mismatches' field. Reads that do not have a field
set will be included. @see http://samtools.sourceforge.net/SAMv1.pdf
Possible operators:
<, <=, =, >, >=, ^^ (flag unset), && (flag set)
Example: (do NOT use reads with mapping quality <= 20, or multiple perfect hits)
-filter 'MAPQ>20' -filter 'H0=1'
----------------------------------------------------------------------------
CODOC 0.0.1 (c) 2013
Usage of the decompressor HEAD command:
Usage: java -jar x.jar decompress HEAD -covFile <arg> [-keepWorkDir] [-tmpDir <arg>] [-v]
Params:
-covFile,--cov <arg> Compressed coverage input file.
-keepWorkDir Do not delete the work directory (e.g., for debugging purposes).
-tmpDir,--temp <arg> Temporary working directory (default is current dir).
-v,--verbose be verbose.
Prints the header from a compressed coverage file.
Usage of the decompressor QUERY command:
Usage: java -jar x.jar decompress QUERY [-chrLen <arg>] -cov <arg> [-keepWorkDir] [-scale <arg>] [-tmpDir <arg>] [-v] [-vcf <arg>]
Params:
-chrLen,--chrLenFile <arg> Chromosome lengths (file format: chr\tlength\n). Use to get proper padding for zero-coverage regions.
-cov,--covFile <arg> Compressed coverage input file.
-keepWorkDir Do not delete the work directory (e.g., for debugging purposes).
-scale,--scaleFactor <arg> Signal scaling factor (default is 1.0).
-tmpDir,--temp <arg> Temporary working directory (default is current dir).
-v,--verbose be verbose.
-vcf,--vcfFile <arg> VCF file used for the compression (optional).
Starts an interactive query session.
Usage of the decompressor TOBED command:
Usage: java -jar x.jar decompress TOBED [-additional <arg>] [-chrLen <arg>] -cov <arg> [-description <arg>] [-keepWorkDir] [-max <arg>] [-min <arg>] [-name
<arg>] -o <arg> [-scale <arg>] [-tmpDir <arg>] [-v] [-vcf <arg>]
Params:
-additional <arg> Additional BED track info (optional).
-chrLen,--chrLenFile <arg> Chromosome lengths (file format: chr\tlength\n). Use to get proper padding for zero-coverage regions.
-cov,--covFile <arg> Compressed coverage input file.
-description <arg> BED description (optional).
-keepWorkDir Do not delete the work directory (e.g., for debugging purposes).
-max <arg> maximum coverage (omit to leave unrestricted).
-min <arg> minimum coverage (omit to leave unrestricted).
-name <arg> BED track name (optional).
-o,--outFile <arg> Output file that will contain the BED intervals.
-scale,--scaleFactor <arg> Signal scaling factor (default is 1.0).
-tmpDir,--temp <arg> Temporary working directory (default is current dir).
-v,--verbose be verbose.
-vcf,--vcfFile <arg> VCF file used for the compression (optional).
Creates a BED file containing regions that are above/below given coverage thresholds.
Usage of the decompressor TOWIG command:
Usage: java -jar x.jar decompress TOWIG [-chrLen <arg>] -cov <arg> [-keepWorkDir] -o <arg> [-scale <arg>] [-tmpDir <arg>] [-v] [-vcf <arg>]
Params:
-chrLen,--chrLenFile <arg> Chromosome lengths (file format: chr\tlength\n). Use to get proper padding for zero-coverage regions.
-cov,--covFile <arg> Compressed coverage input file.
-keepWorkDir Do not delete the work directory (e.g., for debugging purposes).
-o,--outFile <arg> Output file that will contain the rounded approximated coverage values (optional).
-scale,--scaleFactor <arg> Signal scaling factor (default is 1.0).
-tmpDir,--temp <arg> Temporary working directory (default is current dir).
-v,--verbose be verbose.
-vcf,--vcfFile <arg> VCF file used for the compression (optional).
Converts the coverage data to a WIG file.